Arsenic is a non-metal found under 'Group 5' and 'Period 4' of the periodic table of elements. The effects of arsenic on plant and animal systems have been disastrous in agriculture, medicine and warfare (Wood, 1974). The large-scale use of pesticides containing arsenic, for example, has contributed to the environmental contamination by arsenite (AsIII) and arsenate (AsV), the most common oxidation states for soluble arsenic. The oxidation state of arsenic-containing compounds is related to the toxicity of the arsenic-containing molecule. Both forms of arsenic are common contaminants in soil and water.
Arsenite (AsIII), the most toxic form of arsenic, binds sulfur groups of essential cysteines in proteins. This disrupts the three dimensional structure of proteins thereby affecting protein functionality. In cases where such proteins are critical to the survival of the cell, large quantities of arsenite can cause cell death.
Arsenate (AsV) is less toxic than arsenite but is still harmful to cells. Since the structure of arsenate (AsO4-3) is similar to phosphate (PO4-3), arsenate acts as a phosphate analog and competes with the accumulation of adenosine triphosphate (ATP), a primary energy storage form in the cell (Coddington, 1986).
An antidote for arsenite poisoning in humans is British Anti-Lewisite (BAL) (Oehme, 1972). This drug was developed during World War I in response to gas warfare with arsenical compounds. BAL functions by forming a five membered ring complex with arsenite, which, because of its stability, displaces arsenite from cross-linked proteins, thereby decreasing the toxic effects of arsenite.
The bacterium, Alcaligenes faecalis, detoxifies arsenite by converting it to arsenate. The enzyme arsenite oxidase (AO) carries out the reaction in the periplasmic space of the bacterium such that arsenite is converted to arsenate before it can enter the cytoplasm and bind to intracellular proteins. The oxidation of arsenite occurs by transferring two electron equivalents from arsenite to a molybdenum atom in a molybdopterin cofactor in arsenite oxidase (Fig. 1). The electrons are then transferred intramolecularly to a 3Fe-4S cluster and a 2Fe-2S cluster, from where they are donated to azurin and/or cytochrome c protein in the periplasmic space of the cell, and eventually are deposited on oxygen to make water (Anderson et al., 1992).
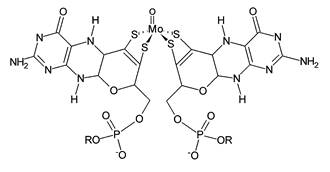
Figure 1. Structure of a molybdopterin cofactor found at the active site of arsenite oxidase. The molybdopterin cofactor has a central molybdenum atom liganded to two pterin rings via dithiolene bridges. R = adenosine monophosphate. The molybdenum atom is MoVI in the oxidized state and is reduced by two electrons to MoIV when arsenite binds (Anderson et al., 1992).
Other redox-active enzymes containing molybdopterin cofactors which catalyze electron transfer reactions have been described (Hille, 1996). However, in many of these proteins, arsenite is a potent inhibitor of activity and has been shown to bind irreversibly within the coordination sphere of the molybdenum atom (George and Bray, 1983). This results in a decrease in redox potential of the molybdopterin cofactor and prevents electron transfer. Since arsenite does not bind irreversibly to arsenite oxidase, a different binding mechanism may be present in this enzyme.
Although arsenite is known to bind sulfur groups in cysteines, these amino acids are unlikely to be the binding site of arsenite in arsenite oxidase since covalent modification of cysteines has no effect on the activity of the enzyme. Histidines, however, have been shown to be essential for activity (McNellis and Anderson, 1998) and may form a binding mechanism for arsenite. This way, arsenite would not be required to bind within the coordination sphere of molybdenum, and may bind loosely enough to dissociate for enzyme turnover.
Three-dimensional structures of other Mo-containing enzymes show that in some enzymes, at least one histidine is located along the tunnel leading from the exterior of the enzyme to the atom. Recent data (Ellis et al., 2001) suggest that similar histidines may be present in arsenite oxidase. These histidines could provide a binding site for arsenite such that the arsenite is positioned close enough to the molybdenum atom to allow electron transfer via orbital overlap. To determine if the histidines putatively involved in arsenite binding are solvent accessible and necessary for enzyme activity, this study involved the modification of solvent accessible histidines with 14C-diethylpyrocarbonate (DEPC) (Miles, 1977), digestion of the protein with trypsin and isolation of the peptides containing the radiolabel by HPLC.
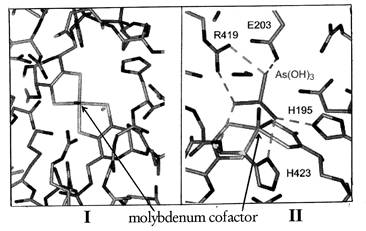
Figure 2. The active site of arsenite oxidase viewed down (I) and across (II) the solvent access channel. The molybdenum center lies at the bottom of a funnel-shaped cavity that provides solvent access to the histidines (H195 and H423) at the active site (Anderson et al., 2001).
Materials and Methods
Arsenite oxidase was purified by published procedures (Anderson et al., 1992). Diethylpyrocarbonate (DEPC), 14C-diethylpyrocarbonate (O(14CO2C2H5)2), guanidine, dithiothreotol (DTT) and iodoacetic acid were obtained from Sigma Chemical Company. Working stock concentrations of DEPC were determined spectroscopically by the reaction with 10mM imidazole at pH 7.5, 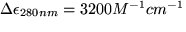 (Miles 1977). A Hewlett Packard 8452 diode array spectrophotometer was used for all assays and spectra.
(Miles 1977). A Hewlett Packard 8452 diode array spectrophotometer was used for all assays and spectra.
The modification of solvent accessible histidines in arsenite oxidase (AO) was carried out by the incubation of 5 nmol of AO with 500 nmol of (14C-)-DEPC in 50mM phosphate buffer (pH 7.0) for 30 minutes at room temperature. Removal of excess (14C-)-DEPC was performed by gel filtration chromatography (G-25 Sephadex) and eluted with 100mM TRIS-HCl at pH 8.4. Fractions containing protein (as evidenced by absorbance at 280 nm) were pooled. The pool was concentrated by microfitration to ~5 uL. Reaction of AO with DEPC was confirmed by the inactivation of the enzyme. Enzyme activity was determined by measuring the rate of reaction of DEPC-modified AO in 50mM MES pH 6.0, 200uM sodium arsenite and 60uM 2,4-dichlorophenolindolphenol (DCPIP) (Anderson et al., 1992). In the control experiment, AO was subjected to the same treatment, except that DEPC was omitted and replaced with an equal volume of anhydrous ethanol.
Following the modification step, the enzyme was denatured in 6M guanidine, treated with 40mM DTT and incubated at room temperature for 30 minutes. The reduced protein was then reacted with 100mM iodoacetic acid for 30 minutes at room temperature. Excess reagents were removed by dialysis against 1000 fold excess volume of 6M guanidine. The chemically modified and denatured AO was concentrated to 10uL via microfitration and then diluted with 10mM TRIS-HCl at pH 8.0 to bring the final concentration of guanidine to < 2.0M.
The modified and denatured protein was digested overnight at room temperature with 80ug/mL trypsin. The resulting peptides were isolated by C18 high-pressure liquid chromatography (HPLC) by application of a linear gradient from 080were made from the radiolabeled fractions based on the radioactive count as determined by broad range counting in a Beckman LS 6000IC scintillation counter using 30% cocktail.
Results and Discussion
(14C-)-diethylpyrocarbonate (O(14CO2C2H5)2) inactivates arsenite oxidase by reacting with all solvent accessible histidyl residues to yield an N-carboxyhistidyl derivative (Fig. 3) which retains the 14C radiolabel throughout the course of all the reactions that follow. Activity assay experiments showed an 80in activity of (14C)-DEPC modified arsenite oxidase suggesting that DEPC had indeed modified essential amino acids in AO (data not shown). Previous published data suggests that these amino acids are histidines (McNellis and Anderson, 1998).
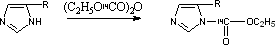
Figure 3. Reaction of 14C-diethylpyrocarbonate with Histidyl Residues.
Treatment of the (14C)-DEPC-modified AO with 6M guanidine caused the protein to unfold, thereby exposing any buried histidine(s) to reaction with unlabeled DEPC. This way, although all histidines were modified by DEPC, only those that were solvent accessible had the radioactive label. To disrupt the three-dimensional structure of AO by reducing the disulfide bonds, dithiothreotol (DTT) was added to the denatured protein. Following this, all exposed thiol groups were blocked by reacting with iodoacetic acid. This step prevented crosslinking of peptides and proteins during further chemical manipulation.
Dialysis of the chemically modified AO against 6M guanidine was deemed to be effective at removing excess iodoacetic acid and DTT as previously described by Fujita et al. (1984). However, the presence of excess 6M guanidine inactivates trypsin (Fujita et al., 1984) . As determined in separate experiments, trypsin was shown to be active if the guanidine concentration was kept below 2M (data not shown).
Although dilution of the denatured AO to 2M guanidine resulted to the precipitation of the protein, smaller peptide fragments were generated overnight by digestion with trypsin as evidenced by the disappearance of the protein precipitate (into soluble peptides) by gel electrophoresis (Kimpler and Anderson, 1999) and the large number of peptides separated by HPLC (Fig. 4). This confirms that trypsin was active and produced peptides from denatured and DEPC-modified arsenite oxidase.
Fractions eluting from the reverse phase chromatography column were tested for radioactivity to locate the peptides containing histidines that were solvent accessible in the native arsenite oxidase. Several peptide fractions contained the radioactive label (Fig. 5) and were separated into seven pools corresponding to the radiolabeled peaks. These fractions were shown to consist of several peptides as evidenced by amino acid sequencing analysis and further purification by microbore C18 HPLC.
Following purification, the amino acid sequence of each radiolabeled peptide will be determined. Comparison of the peptidyl sequence to that of the X-ray crystallographic structure (Ellis et al., 2001) should indicate if the histidines located close to the molybdenum atom are possible binding sites for arsenite.
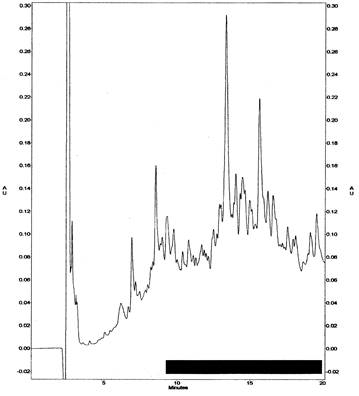
Figure 4. C18 HPLC chromatogram of trypsin digested DEPC-(14C and cold) modified arsenite oxidase. Peptides were separated with a linear gradient from 0% to 80% acetonitrile in 0.1% trifluoroacetic acid over 40 minutes at 1.0mL per minute. Peptides were detected in line by absorbance at 220 nm (ordinate of plot). Fractions were collected at 0.5-minute intervals. The shaded region represents the range of pools made from the fractions containing the most 14C counts (see 5).
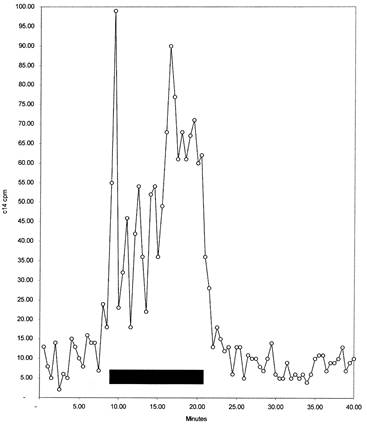
Figure 5. Radioactive 14C elution of digested DEPC-modified arsenite oxidase from C18 HPLC. Fractions were obtained as described in Figure 4. Aliquots from each fraction were taken and the amount of radioactive 14C (in dpm) was determined for each 0.5-mL fraction. The shaded region represents the range of pools made from the fractions containing the most 14C counts.
Conclusion
The bacterium, Alcaligenes faecalis, uses an oxomolybdenum enzyme, arsenite oxidase, to detoxify arsenite by converting it to arsenate via a two-electron transfer reaction. The mode of action of previously studied molybdenum-containing hydroxylases eliminates the possibility of direct electron transfer from arsenite to the Mo atom because an irreversible bond would form and thereby inhibiting the enzyme (Hille, 1996). An alternative mechanism of arsenite oxidase is consistent with the crystal structure of the enzyme (Ellis et al., 2001) whereby at least two histidines are located at the active site of the enzyme in such a way that they could provide a binding site for arsenite and position it to interact with the molybdenum atom through orbital overlap. In this study, the peptides containing the putative histidines were isolated and will be further analyzed.
In the environment, arsenite contamination could theoretically be treated by bioremediation in which bacteria would be used to detoxify arsenite in soil or water. Two disadvantages are associated with this approach: Since bacteria are living cells, they survive only within a narrow set of conditions suitable for growth (appropriate pH, other nutrients, and lack of other toxins). In addition, some bacteria survive in arsenic by oxidizing arsenite to arsenate, while other bacteria survive by converting arsenate to arsenite. A futile cycle is thus set up so that arsenite and arsenate levels may reach an equilibrium, but both forms are continuously present in the environment.
Another approach to environmental decontamination is to design potential arsenite detoxifying chemicals. Such chemicals would be used on a large scale and their functionality would not be affected by environmental factors. However, to design this type of chemical approach, a better understanding of the enzymatic mechanism used by arseniteresistant bacteria is required.
Acknowledgements
The writers thank the IUSB SMART program for providing funds for this research. Additional funding was obtained from IUSB faculty research grants, and from the Petroleum Research Fund.


